RNA-Seq 和蛋白质结构预测是现代生物学研究的重要工具,有助于深入了解疾病的分子机制和潜在疗法的开发。RNA-Seq 是一种分析基因表达的技术,使研究人员能够更好地了解基因调控和基因之间复杂的相互作用。另一方面,蛋白质结构预测提供有关蛋白质功能以及与其他分子相互作用的信息。这对于药物开发非常有用,并且能够识别靶标结合位点并优化候选药物。
挑战
处理这些技术生成的大量数据的复杂性给研究人员带来了巨大的挑战。RNA-Seq 和蛋白质结构预测需要大量的计算和存储开销才能进行准确分析。许多蛋白质的大小和复杂的结合动力学使蛋白质结构预测变得更加复杂,这使得准确预测变得困难。此外,许多蛋白质缺乏真实的结构数据使得验证计算预测变得困难,进一步增加了任务的复杂性。应对这些挑战需要可扩展且可重复的分析方法来获得准确可靠的结果。
解决方案
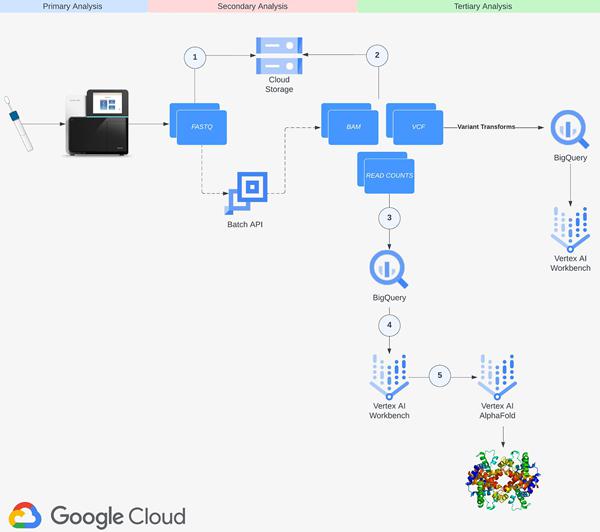
机器学习和云计算的最新进展为应对这些挑战开辟了新途径。AlphaFold是DeepMind开发的深度学习程序,可以根据输入的氨基酸序列准确预测蛋白质的 3D 结构以及与其他分子的结合动力学。Nextflow,一个开源工作流程管理系统和Google Batch是一项完全托管的服务,用于在云计算资源上调度、排队和执行批处理作业,使用 Docker 容器实现基因组数据的可扩展和可重复分析。借助 Google Cloud 的强大功能,我们开发了用于 RNA-Seq 和蛋白质结构预测的端到端管道,利用 BigQuery 和 Vertex AI 高效处理 TB 级数据。通过分享我们的经验,我们希望能够深入了解如何利用 Google Cloud 应对现代生物学和医学中的计算挑战,最终为新发现和创新铺平道路。
方法
第 1 步:设置
创建 Cloud Storage 存储分区。请参阅有关创建存储桶的文档。在项目中启用ArtifactRegistry、Batch、Dataflow、Cloud Life Sciences、Compute Engine、Notebooks、Cloud Storage 和 Vertex AI API。创建服务帐户并在项目级别添加 Cloud Life Sciences Workflows Runner、服务帐户用户、服务使用使用者和存储对象管理员角色。请参阅有关创建服务帐户的文档。
第二步:数据收集
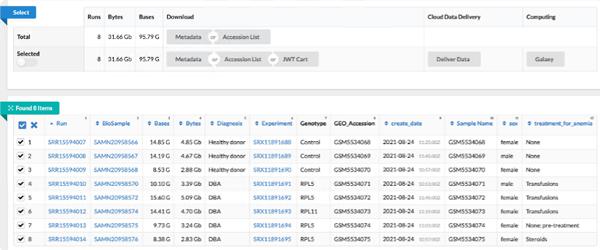
我们解决方案的下一步涉及从公共 NCBI 数据集 ( GSE181830 ) 收集数据,该数据集源自题为“人类骨髓祖细胞的单细胞分析揭示 Diamond-Blackfan 贫血中红细胞生成失败的机制”的论文。研究中使用的患者元数据和 FASTQ 文件(即 *.fq.gz)可通过 NCBI 的 SRA 运行选择器服务下载,并且可以通过向指定存储桶上的 NCBI 服务帐户授予临时存储对象管理员访问权限来直接传送到云存储。数据传输完成后,将发送一封确认电子邮件,指定的存储桶应包含选择传输的文件。或者,您可以从欧洲核苷酸档案馆 ( ENA)下载 FASTQ 文件)网站,方法是输入每次运行的 SRA ID,然后使用 wget 或 curl 将文件下载到本地目录,然后使用gsutil将它们复制到云存储桶。
第 3 步:构建基因组索引
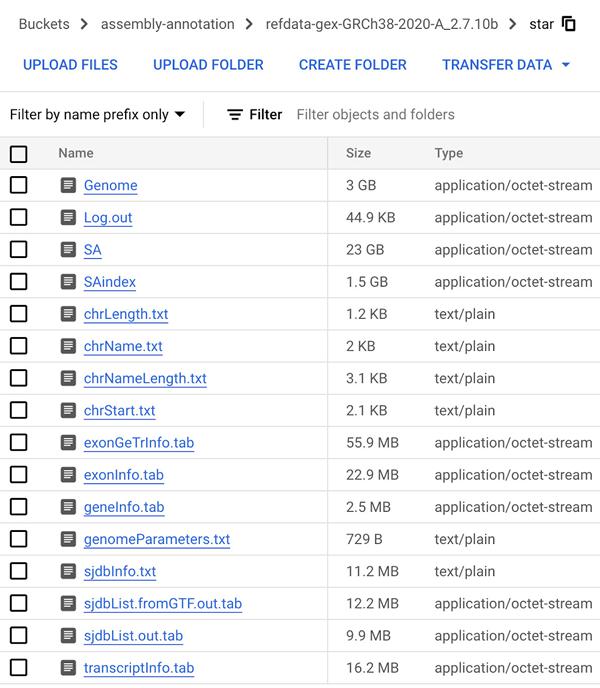
下一步涉及创建基因组索引 (GRCh38.v42),该索引将用于在 RNA-Seq 分析的比对步骤中映射读数。使用从Gencode下载并上传到 Cloud Storage 的转录本和注释文件,我们使用可在 Github 上下载的开源STAR RNA对齐器库 (2.7.10c) 在 Vertex AI Workbench 用户管理的笔记本中构建基因组索引。请参阅有关创建用户管理的笔记本实例的文档。使用指定的输出目录创建基因组索引的过程大约需要一个小时。完成后,我们将输出目录上传到步骤 1 中创建的 Cloud Storage 存储桶。
步骤 4:构建容器镜像并将其上传到 ArtifactRegistry
下一步涉及构建 Docker 容器映像并将其上传到ArtifactRegistry(Google Cloud 的工件存储库)。Nextflow 在您指定的容器中执行管道中的每个步骤。我们需要构建管道将使用的映像,并加载依赖项和库,包括 fastqc、multiqc、rna-star、rsem 以及管道中将引用的 BigQuery Python 客户端库。请参阅有关创建存储库以及将映像推送和拉取到 Artifact Registry 的文档。创建映像并将其推送到存储库后,将 Artifact Registry Reader 角色授予在步骤 1 中创建的服务帐户。我们在工作流程中使用的 Dockerfile 可以在 Github 中查看。
第 5 步:为基因和亚型表达数据创建 BigQuery 数据集
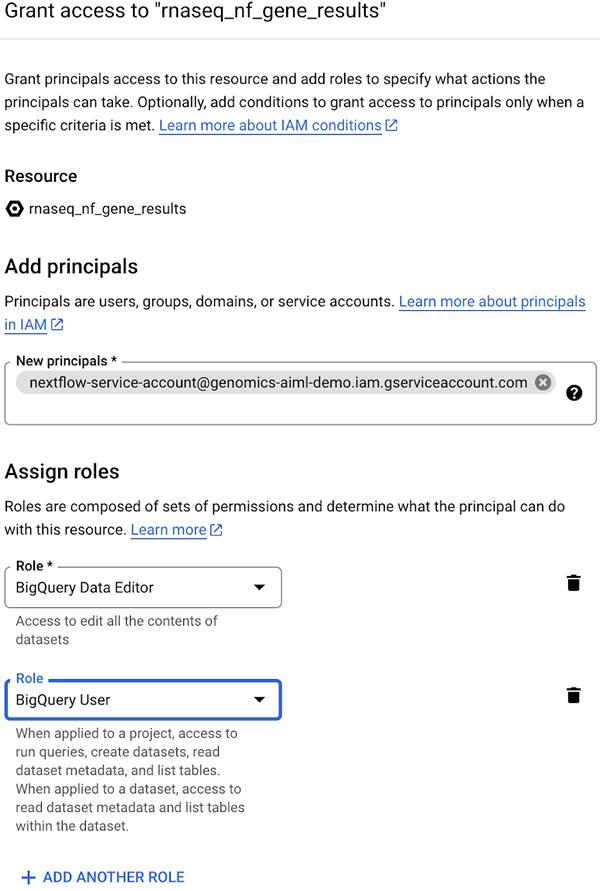
下一步是创建 BigQuery 数据集,其中分别包含基因和异构体读取计数数据。RNA-Seq 管道的最后一步将读取计数数据写入 BigQuery 表,并且数据集必须预先存在。请参阅有关在 BigQuery 中创建数据集的文档。将第一个创建的数据集命名为rnaseq_nf_gene_results,将第二个数据集命名为rnaseq_nf_isoform_results。在这两个数据集中,将 BigQuery Editor 和 BigQuery User 角色授予在步骤 1 中创建的服务帐号。
步骤 6:配置 Nextflow 管道
下一步是配置 Nextflow 管道来执行我们的双端 RNA-Seq 预处理工作流程。这涉及在 Github 存储库中创建 nextflow.config 和 main.nf 文件。我们在工作流程中使用的 nextflow.config、main.nf 和 load_rsem_results_into_bq.py(由 main.nf 引用)文件可以在Github中查看。请参阅有关 Nextflow 脚本编写的文档。使用您的项目和存储桶详细信息更新 nextflow.config 脚本。main.nf 脚本中详细说明的流程步骤为:(1) 使用Trim Galor e 进行接头和质量修整,(2) 使用FastQC进行质量控制读数,(3) 使用RSEM对基因和亚型表达进行比对和估计,以及(4) 将表达式数据写入大查询。
步骤 7:在 Nextflow Tower 上部署管道
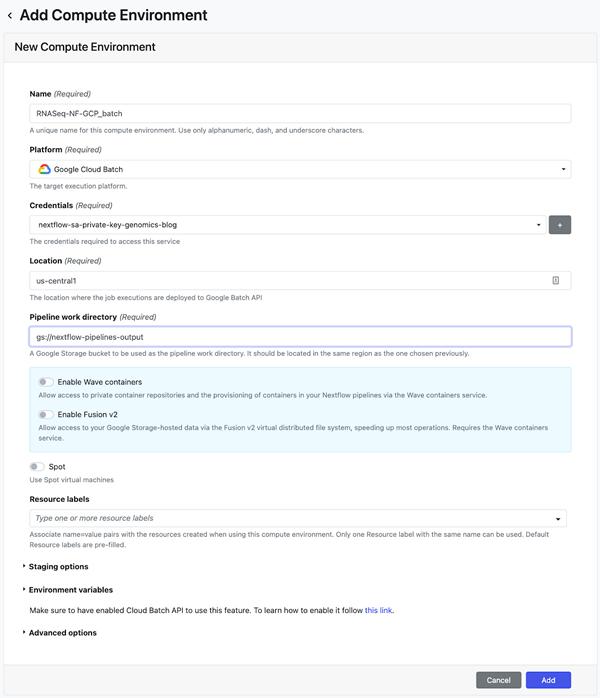
下一步是在 Nextflow Tower上部署管道。Tower 提供对 Google 项目进行身份验证的支持,其中管道将通过服务帐户密钥运行。请参阅有关连接服务帐户密钥的文档。Tower 还支持使用访问令牌连接到托管 Nextflow 配置的 Git 存储库。查看文档连接 Tower 中的存储库。配置服务帐户和存储库凭据后,下一步是创建计算环境。在“添加计算环境”视图中,输入您选择的环境名称(例如 RNASeq-NF-GCP),然后在“平台”下拉列表中选择“Google Batch”。然后,输入将创建工作负载的区域(可以使用步骤 6 中的 nextflow.config 中指定的相同区域)并输入管道工作目录(即 GCS 路径)。
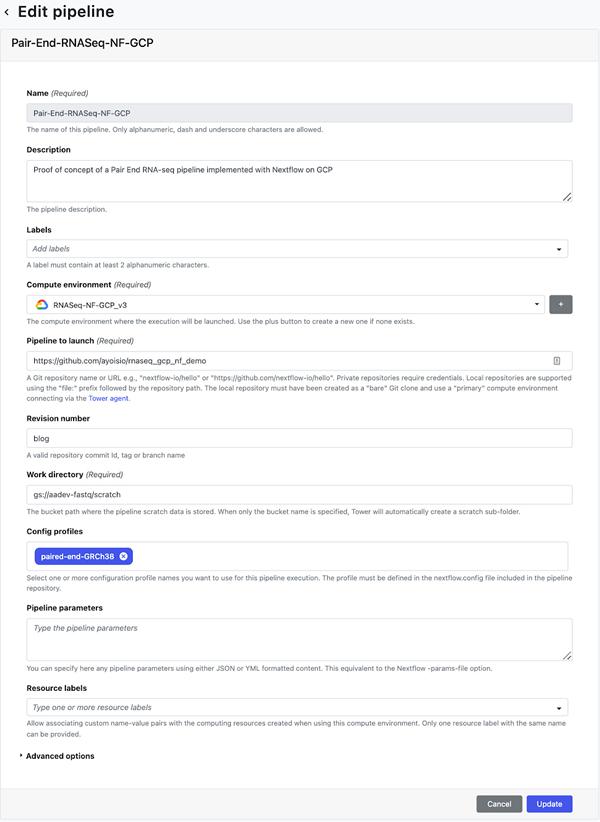
创建环境后,请转到启动板页面上的添加管道视图。在“添加管道”视图中,输入您选择的管道名称,选择上面创建的计算环境,输入托管在步骤 6 中创建的 Nextflow 配置的 Git 存储库 URL,然后输入要在执行期间使用的修订号(即分支)。最后,输入paired-end-GRCh38作为nextflow.config脚本中定义的配置文件。
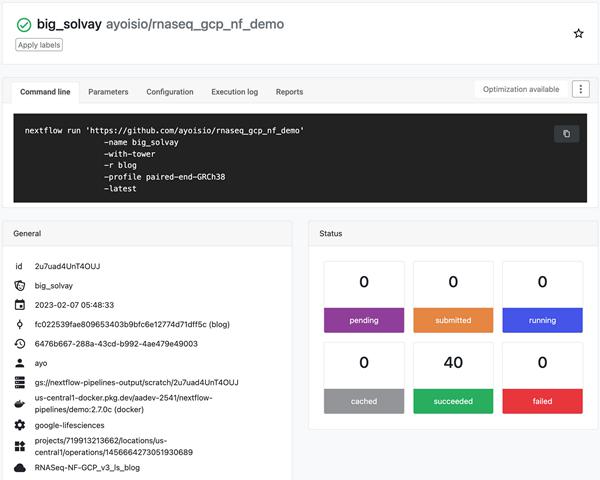
创建管道后,启动它。管道应在后台启动并需要 4-6 小时。您可以通过转到“运行”选项卡并单击“运行 ID”来监视管道的进度。管道完成后,基因组和异构体表达结果将在步骤 5 中创建的基因组和异构体表中可用并可查询。FastQC 报告将在 nextflow 配置中指定的输出目录中提供。
第 8 步:运行 DESeq2 笔记本分析
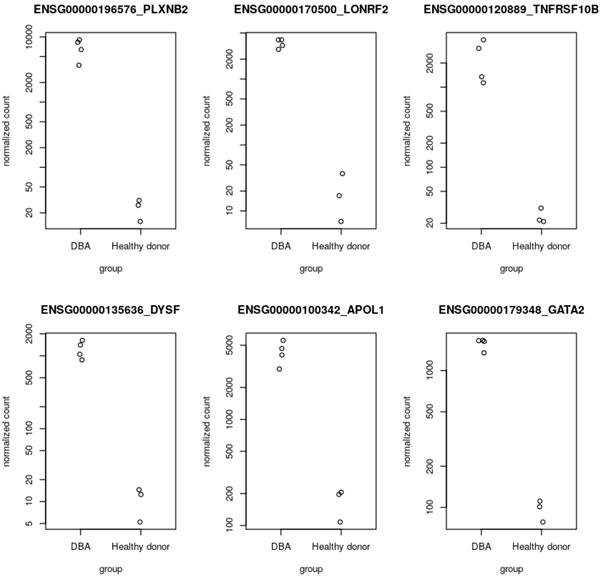
下一步是使用步骤 7 中生成的 BigQuery 中的基因读取表达数据在用户管理的 R 笔记本中运行 DESeq2 分析。DESeq2 是一个 R 包,提供统计方法来检测两个或多个生物之间的差异基因表达。状况。在我们的工作流程中,我们使用 DESeq2 检测 Diamond Blackfan 贫血患者和健康对照之间的差异表达 (DE) 基因。我们使用 AlphaFold 在步骤 10 中可视化顶级 DE 基因。请参阅Github 中的笔记本以供参考。DESeq2 分析输出的几个顶级 DE 基因,包括GATA2、DYSF和TNFRSF10B,在基因组文献中被引用为 Diamond Blackfan 贫血的特征,并验证步骤 1-8 中执行的 RNA-Seq 管道预处理工作流程和笔记本分析。
第 9 步:创建将转录本转换为氨基酸序列的远程函数
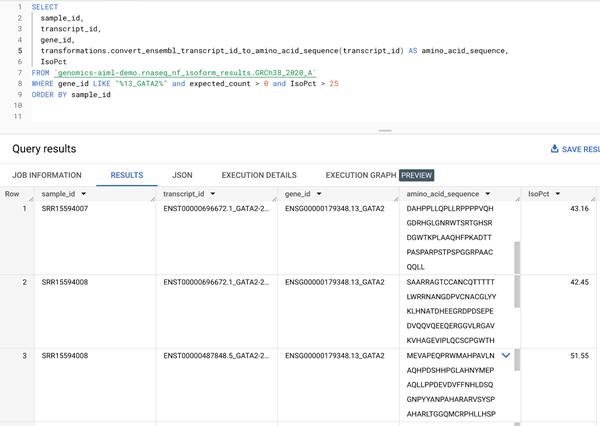
下一步是创建一个远程函数,将基因转录本转换为氨基酸序列。步骤 7 中创建的异构体表达表包括转录本 ID 列,其中包含每个基因的异构体表达水平。我们将转录本 ID 列传递给远程函数,该函数调用 Ensembl Sequence REST API并在给定输入基因转录本的情况下输出氨基酸序列。由于 AlphaFold 接受氨基酸序列作为输入,因此这提供了一种可扩展的方式来生成氨基酸序列,这些氨基酸序列将直接从我们的 RNA-Seq 管道生成的表达数据提供给 AlphaFold。请参阅有关创建远程函数的文档。查看代码在Github上供参考。在我们的工作流程中,我们生成对应于步骤 8 中 DE 基因输出的转录本的氨基酸序列。创建 GATA2 转录本序列的示例查询如下所示。
第 10 步:使用 AlphaFold 可视化氨基酸序列
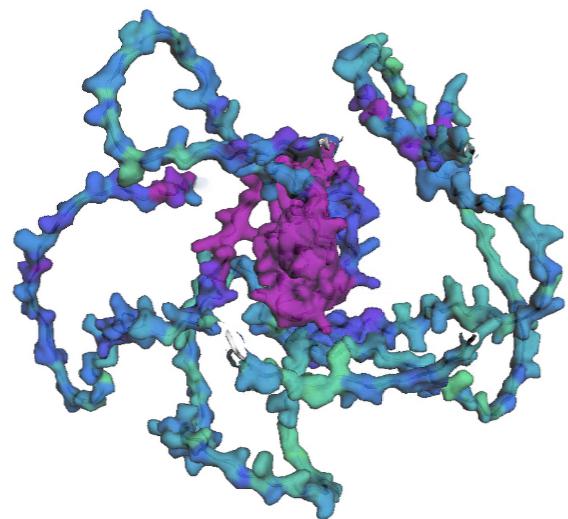
最后一步是使用 AlphaFold 可视化氨基酸序列。Google Cloud 在 ArtifactRegistry 中提供了一个自定义的 Docker 映像,并带有预安装的软件包,用于在 Vertex AI Workbench 中启动笔记本实例并运行 AlphaFold。您可以在Vertex AI 社区内容中找到笔记本、Dockerfile 和构建脚本。请参阅相关博客文章,了解有关使用推荐的 GPU 加速器部署笔记本电脑的详细说明。使用 BigQuery Python 客户端,我们执行查询以生成 DE 基因的氨基酸序列,然后将其传递并通过 AlphaFold 运行。DE 基因 GATA2 之一的蛋白质结构预测示例如下所示。
下一步
扩展的工作流程将包括在 Nextflow 上实施并行变体调用管道、生成 VCF 文件并将其上传到 Cloud Storage,以及使用变体转换工具将变体数据加载到 BigQuery 中。然后,来自 RNA-Seq 工作流程的异构体表达数据将与变体数据相结合,生成每个样本特有的氨基酸并包含已识别的变体。这将为精准医学指导方法提供额外支持。请参阅Github中的variant-calling.nf 脚本,了解参考变体调用管道。
在本博客中,我们开发了一个用于 RNA 测序和蛋白质结构预测的端到端管道,它以可扩展的方式利用 BigQuery 和 Vertex AI 来处理基因组数据。我们希望我们能够就如何利用 Google Cloud 应对现代生物学和医学中的计算挑战提供见解,为新发现和创新铺平道路。